
Gambar:Phylogenetic tree of coronaviruses.jpg
Euweuh résolusi nu leuwih luhur.
Phylogenetic_tree_of_coronaviruses.jpg (571 × 451 piksel, ukuran koropak: 75 KB, tipeu MIME: image/jpeg)
| Berkas ieu asalna ti Wikimedia Commons. Kaca ngeunaan katerangan berkas ieu di Commons aya disalin di handap. Commons téh hiji gudang berkas bébas nu eusina disumbang ku balaréa. |
{kind=link}
Ringkesna
| Pedaran |
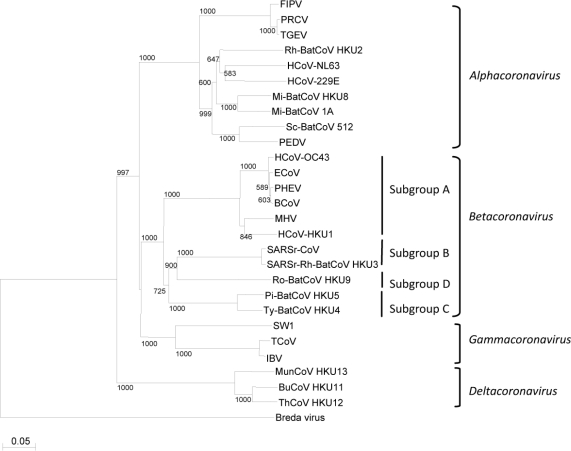
English: Phylogenetic analysis of RNA-dependent RNA polymerases (Pol) of coronaviruses with complete genome sequences available. The tree was constructed by the neighbor-joining method and rooted using Breda virus polyprotein. Bootstrap values were calculated from 1000 trees. 1118 amino acid positions in Pol were included. The scale bar indicates the estimated number of substitutions per 20 amino acids. All abbreviations for the coronaviruses were the same as those in Figure 1. |
| Titimangsa | |
| Sumber | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ |
| Pangarang | Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen |
Lisénsi
Berkas ini berlisensi di bawah lisensi Creative Commons Atribusi 3.0 Tanpa Adaptasi
- Anjeun bébas:
- pikeun babagi – pikeun nyalin, nyebarkeun, sarta nyalabarkeun karyana
- pikeun nyampurkeun – pikeun ngarobah karyana
- Dumasar kana kaayaan di handap:
- atribusi – Anjeun kudu ngatribusi karya ku cara nu geus ditangtukeun ku nu nyieun atawa nu méré lisénsi (tapi lain ku jalan nu sigana téh maksa pikeun Anjeun maké éta karya).
Jujutan berkas
Klik dina titimangsa pikeun nempo koropak nu aya dina mangsa éta.
| Titimangsa | Miniatur | Ukuran | Pamaké | Kamandang | |
|---|---|---|---|---|---|
| kiwari | 7 Maret 2020 02.42 | | 571 × 451 (75 KB) | Guest2625 | Uploaded a work by Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ with UploadWizard |
Pamakéan berkas
Ieu berkas dipaké ku kaca di handap:
Pamakéan berkas sajagat
Wiki lianna anu maké ieu berkas:
- Pamakéan di bn.wikipedia.org
- Pamakéan di en.wikipedia.org
- Pamakéan di fa.wikipedia.org
- Pamakéan di fr.wikipedia.org
- Pamakéan di ig.wikipedia.org
- Pamakéan di sq.wikipedia.org
{kind=link}